Spatial TME analysis
Spatial-PHLEX is the final submodule of TRACERx-PHLEX. It is a nextflow-based pipeline for the analysis of spatially resolved single cell data. It is designed for the highly granular phenotypic outputs of TYPEx, but owing to its simple input format can be used with any single cell coordinate data where single cells are assigned a phenotype or class label with minimal effort.
Workflow options
- Spatial-PHLEX provides three primary workflow options for spatial cell data:
Workflow 1: Spatial clustering

Overview of the Spatial-PHLEX spatial clustering workflow.
Workflow 2: Cellular barrier scoring
Definition of a barrier
The Spatial-PHLEX module allows the user to select a trio of cell types on which to perform barrier scoring, interrogating the extent to which a barrier cell type is spatially inter-positioned between a second and third cell type, the latter clustered by the DB-Scan spatial clustering method also invoked in Spatial-PHLEX. Six different barrier scores are automatically output by Spatial-PHLEX.
Using the example of a fibroblast cell type as the barrier cell type between CD8 T cells and tumour cell spatial clusters, each metric responds to a slightly different question:
Barrier content- How many fibroblasts are typically spatially inter-positioned between a CD8 T cell and tumour nest?
**Binary barrier **- How often is a fibroblast spatially inter-positioned between a CD8 T cell and tumour nest on at least one path from CD8 T cell to tumour nest?
Binary adjacent barrier - How often is a fibroblast spatially inter-positioned between a CD8 T cell and tumour nest and positioned at the tumour-stroma interface, on at least one path from CD8 T cell to tumour nest?
Weighted barrier - To what extent are fibroblasts spatially inter-positioned between CD8 T cells and tumour and positioned in the vicinity of the tumour nest?
All-paths barrier fraction - How often is a fibroblast spatially inter-positioned between a CD8 T cell and tumour nest, accounting for all possible routes from CD8 T cell to tumour nest?
All-paths adjacent barrier fraction - How often is a fibroblast spatially inter-positioned between a CD8 T cell and tumour nest and positioned at the tumour-stroma interface, accounting for all possible routes from CD8 T cell to tumour nest?
Workflow 3: Barrier scoring to spatial clusters
Example usage
First, clone the repository. Then, navigate to the Spatial-PHLEX directory.
git clone https://github.com/FrancisCrickInstitute/Spatial-PHLEX
cd Spatial-PHLEX
Then load Nextflow and Singularity on your system with e.g. (on SLURM):
ml Nextflow/22.04.0
ml Singularity/3.6.4
Set the Singularity cache directory environment variable, to store the container image:
export NXF_SINGULARITY_CACHEDIR='./singularity'
The Spatial PHLEX pipeline can then be run with the following command:
nextflow run ./main.nf \
--workflow_name 'clustered_barrier' \
--objects "$PWD/data/PHLEX_test_cell_objects.txt"\
--objects_delimiter "\t" \
--image_id_col "imagename"\
--phenotyping_column 'majorType'\
--phenotype_to_cluster 'Epithelial cells'\
--x_coord_col "centerX"\
--y_coord_col "centerY"\
--barrier_phenotyping_column "majorType" \
--barrier_source_cell_type "CD8 T cells"\
--barrier_target_cell_type "Epithelial cells"\
--barrier_cell_type "aSMA+ cells"\
--n_neighbours 10\
--outdir "../results" \
--release 'PHLEX_test' \
--plot_palette "$PWD/assets/PHLEX_test_palette.json" \
-w "./scratch"\
-profile {your_nf-core_profile} \
-resume
Replace {your_nf-core_profile} with the profile for your organisation, e.g. crick. For more information on nf-core profiles, see the [list of available nf-core configs](https://github.com/nf-core/configs) plus other advice on pipeline configuration [here](https://nf-co.re/usage/configuration#profiles).
Input Files
Required Inputs
- cell_objects.csv
A plaintext, delimited file containing single cell-level coordinate data for a set of images, plus their phenotypic identities.
- metadata.csv
Optional. A plaintext, delimited file containing metadata information about the images in cell_objects.csv. To run the pipeline this file must contain, for each image, an image identifier (e.g. ‘imagename’ specified with the flag –image_id_col), and the width and height in pixels for every image as columns with the header ‘image_width’ and ‘image_height’. If this file is not provided, the pipeline will attempt to infer approximate image dimensions from the maximum x,y cell coordinates for each image from the cell_objects.csv file.

The Spatial PHLEX input dataframe has a simple format allowing cell coordinate results from other imaging modalities to be processed with Spatial-PHLEX.
Outputs
Cell type specific spatial clusters
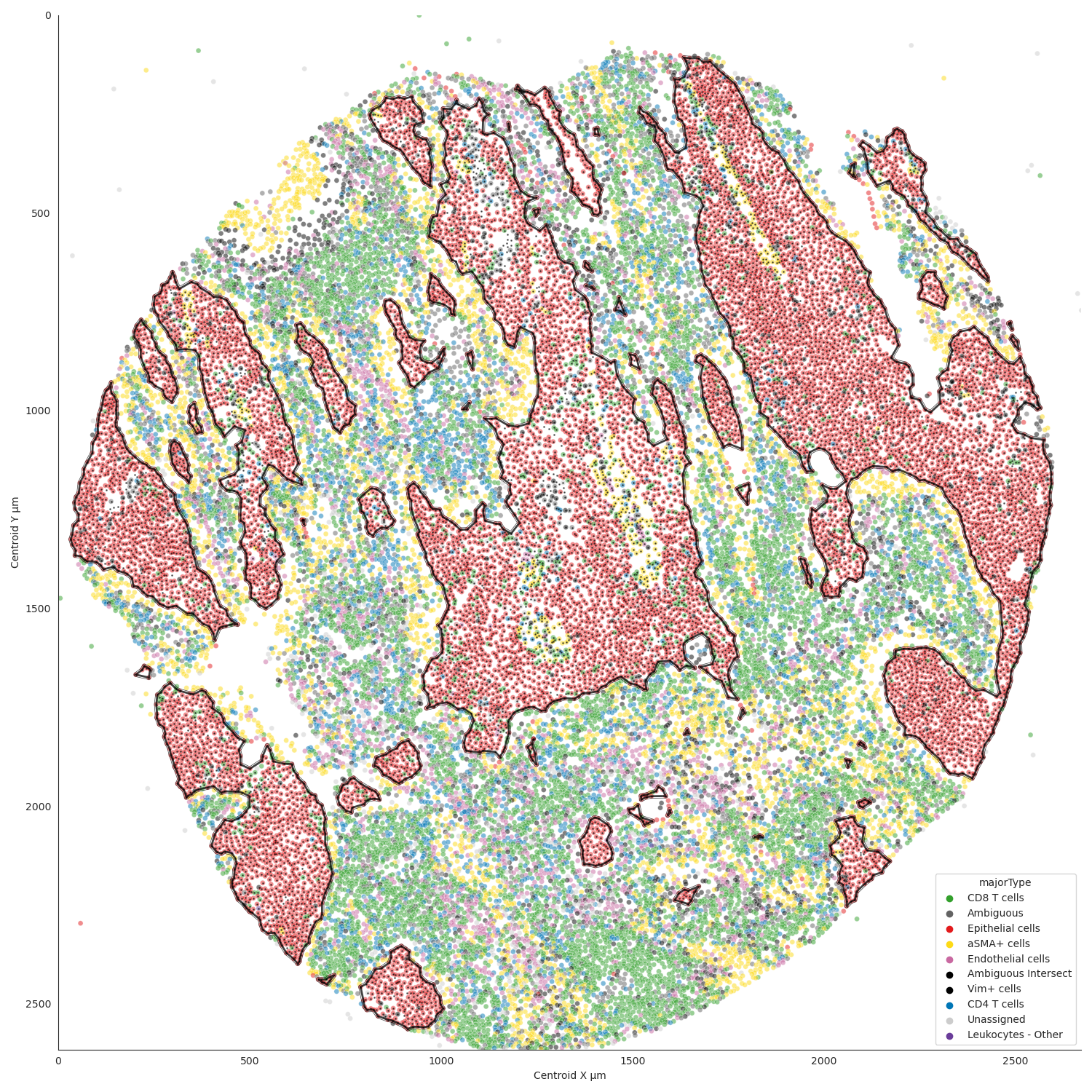
Example spatial cluster plot produced with Spatial-PHLEX.
Intracluster densities
Barrier scores
Output from Spatial-PHLEX has the following directory structure.
results
├── graph
│ ├── aggregated_barrier_scoring
│ └── raw_barrier_scoring
└── spatial_clustering
└── pipeline_info
Note
The name of the raw_barrier_scoring directory will vary depending on which Spatial-PHLEX workflow is specified.
Parameters
Spatial PHLEX parameters are defined in the nextflow.config file in the Spatial PHLEX base directory.
Spatial-PHLEX param |
Definition |
Input options |
|---|---|---|
barrier_cell_type |
The type of cell forming the barrier in the barrier scoring calculation. |
e.g. Myofibroblasts |
barrier_phenotyping_column |
Column name in the objects table used to derive cell types for barrier scoring. Can be distinct from the phenotyping_column specified for spatial clustering if multiple phenotypic columns exist in the file. |
e.g. cellType, phenotype |
barrier_source_cell_type |
The source cell type to compute barrier scores for. |
e.g. `’CD8 T cells’ ` |
barrier_target_cell_type |
The target cell type to compute barrier scores for. |
e.g. ‘Epithelial cells’ |
dev |
Development mode; sample a subset of input images. |
true, false |
graph_type |
The method of graph construction from cell positional data. |
‘nearest_neighbour’ (default),’neighbourhood’,’spatial_neighbours’ |
metadata |
(optional) Path to the metadata file containing width and height information about images to be analysed. Supplying this file allows plots to have exact dimensions of input images. |
e.g. ‘/path/to/metadata.txt’ |
metadata_delimiter |
Delimiter of the metadata file. |
e.g. ‘t’ |
n_neighbours |
Number of nearest neighbours for nearest_neighbour graph construction. |
10 |
neighborhood_input |
globbable path to csv files containing neighbouRhood output produce by CellProfiler module. |
e.g. ‘/path/to/results/segmentation///neighbourhood.csv’ |
neighbourhood_module_no |
Module number of the neighbouRhood proces sin the CellProfiler pipeline |
e.g. 865 |
number_of_inputs |
Number of images to process the data for in development mode. |
2 |
objects |
Path to the cell objects dataframe. |
e.g. ‘/path/to/objects.csv’ |
objects_delimiter |
Character delimiting the objects dataframe. |
e.g. ‘t’ |
outdir |
Root output directory where results will be created. |
../../results |
overwrite |
Overwrite results published to the results directory, if they already exist. |
true |
phenotyping_column |
The column name in the objects dataframe defining the phenotypes of the cells. |
e.g. ‘cellType’; ‘Ki-67+ve’ |
publish_dir_mode |
Way Nextflow generates output in the publish directory. |
default: ‘copy’ |
release |
Release directory. Identifier for the data analysis run. |
e.g. ‘2022-08-23’ |
workflow_name |
Spatial PHLEX workflow to run on the data. |
Options: ‘clustered_barrier’, ‘default’,’spatial_clustering’, ‘graph_barrier’ |
phenotype_to_cluster |
The type of cells in the phenotyping_column to perform spatial clustering on. |
Options: ‘all’ (for all types) or ‘Epithelial cells’ etc |
image_id_col |
Column name specifying the image that a given cell pertains to. |
e.g. ‘imagename’, ‘Image_ID’ |
x_coord_col |
Column header of x coordinate data. |
‘centerX’, ‘x’ etc |
y_coord_col |
Column header of y coordinate data. |
‘centerY’, ‘y’ etc |
plot_palette |
(optional) Path to a json-formatted custom palette file to use for spatial clustering output plots. Must have a hex entry for all phenotypes in the phenotyping_column used for spatial clustering. See PHLEX_test_palette.json in the Spatial-PHLEX assets directory. If you do not have a custom palette choose ‘default’. |
‘default’ or /path/to/palette.json |
Troubleshooting
Input file paths are not found
This may be caused by several issues:
1. The input file path is incorrectly specified in the Spatial-PHLEX run command. Check all input file paths are correct.
2. If input file paths are correct for your system then it is possible that Spatial-PHLEX is not able to access the input files due to the file permissions. Check that the user running the Spatial-PHLEX pipeline has read access to the input files.
3. If file paths are correct, and permissions are correct, then Spatial-PHLEX may not be mounting the input file paths correctly in the Spatial-PHLEX Singularity container. Spatial-PHLEX tries to mount filepaths automatically, but occasionally this may fail. In this case the --singularity_bind_path parameter can be specified manually in the Spatial-PHLEX run command or nextflow.config file. Multiple bind paths may be specified simultaneously, separated by a comma e.g. --singularity_bind_path "/path/to/bind1,/path/to/bind2".
Cell type niche analysis via density-based spatial clustering
Some information.
Cellular barrier scoring
Some more information.